Bakkeren
Plant Pathology Lab - Ustilago Research
Jump to 'Current research
projects'
BACKGROUND
Smut Fungi
Smut fungi are basidiomycete
fungi and belong to the order of the Ustilaginales. They are named
after the very conspicuous symptoms, black masses of teliospores resembling
soot or smut, which they often produce on host plants. Although controlled in
our Western societies with fungicides and in some cases resistant crops, smut
diseases remain very important plant pathogens world-wide. Indeed, the
potential to cause epidemics is lurking even in Western societies, especially
with the deregulation of many fungicides.
Over 1,100 smut species have been recognized in over 50
genera infecting more than 4,000 plant species belonging to approximately 75
different plant families. The biggest group infects monocots belonging to the
Gramineae which include cereal crops. The smuts encompass among others, the
genera Ustilago and Tilletia,
the latter causing so-called bunt diseases of various cereal crops.
Phylogeny
We have published a preliminary (Bakkeren
et al., 2000) and a more extensive study (Menzies
et al., 2003) on phylogenetic
relationships between some of the small grain-infecting smut and bunt fungi
belonging to the Ustilaginomycetes and pointed to an intriguing correlation
with a phylogenetic tree of their respective host plants. In the near future we
will see a more extensive study from our collaborators.
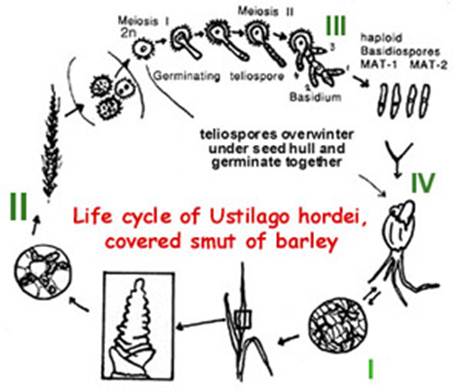
Life cycle
Ustilago hordei is
representative of a group of fungal pathogens that cause economically important
smut diseases
world-wide on small grain cereals. These fungi grow as dikaryotic hyphae within
developing seedlings, residing mainly in the meristem, without causing dramatic
symptoms (I, diagram below). Upon flowering, the fungal cells proliferate, form
thick walls and karyogamy takes place, upon which masses of dark, echinulated
teliospores are formed that replace the developing seeds (II). Teliospores are
survival structures. They can disseminate and survive often under the seed hull
of healthy barley seeds. Under the right conditions both seed and teliospores
will germinate. Diploid teliospores germinate and undergo meiosis to yield
haploid cells which are called basidiospores (III). These meiotic progeny must
mate and form a dikaryotic hypha to re-infect the host. The fungus can then
enter the emerging seedling by direct penetration (IV; Hu et al, 2002). Infection is a
prerequisite for completion of the sexual phase of the life cycle, i.e., the
formation of teliospores. Sex and pathogenicity are therefore interconnected in
U. hordei and related smut fungi, and the mating-type genes are
considered pathogenicity factors.
fungal
proliferation | infection process
Vital statistics
HIGHER FUNGI
Subdivision: BASIDIOMYCOTINA
Class: HEMIBASIDIOMYCETES (TELIOMYCETIDAE)
Order: Ustilaginales
Genus: Ustilago
Species: hordei
|
covered smut |
||
|
Disease |
Main
hosts |
Pathogen |
|
covered smut |
barley & oat (Hordeum vulgare
& Secale, but also Elymus & Agropyron spp.) |
Ustilago hordei (suggested nomenclature: Ustilago segetum f.sp.
hordei) |
Genome
size of U. hordei is approx. 25 Mbp; 250 BAC clones for 1x genome
coverage (Bakkeren et al. 2006. Physical mapping of the genome of the fungal
pathogen Ustilago hordei and
annotation of the 500 kb MAT-1 sequence. Fungal Genet Biol, 43, 655-666)
Mating systems
U. hordei has a
bipolar mating system controlled by one mating-type locus (MAT) and two
alleles or alternative specificities exist for the locus, MAT-1 and MAT-2.
Hybridization experiments with the well-characterized a and b mating-type
genes from the related species, Ustilago maydis, revealed that U.
hordei possesses similar mating-type functions located at so-called a
and b gene complexes within the MAT locus (Bakkeren
et al., 1992). In contrast to U.
hordei, U. maydis has a tetrapolar mating system due to the fact
that the a and b loci are on separate chromosomes and therefore
segregate independently during meiosis. In U. maydis, two specificities
exist for the a locus: a1 and a2. The a locus encodes
cell-type specific pheromones (mfa) as well as the pheromone receptors (pra)
that recognize pheromones from compatible mating partners. The b locus
of U. maydis is multiallelic and contains two divergently transcribed
genes, bE (bEast) and bW (bWest). The two genes
have similar organizations in that each encodes a variable amino-terminal
region, a conserved carboxy-terminal region and an intervening
homeodomain-related motif. The b locus controls pathogenicity and
completion of the life cycle through self versus non-self recognition between
bE and bW polypeptides to establish a regulatory factor. The homologues of the a
and b genes from U. maydis and U. hordei have been
demonstrated to be conserved in structure and function (Bakkeren
and Kronstad, 1993; Bakkeren
and Kronstad, 1996). In both species, only cells of opposite
mating-type, that is, having different specificities at both a and b,
successfully mate and form colonies with aerial hyphae (Fuz+ reaction).
These combinations are infectious when inoculated into host plants. Conversely,
haploid strains or incompatible partners of the same mating type form
yeast-like colonies and are non-infectious. These yeast-like cells divide by
budding and are amenable to many molecular techniques such as transformation
and gene replacement.

We
have shown that a and b are physically linked on the largest
chromosome of U. hordei and together encode key functions within the MAT
locus. Preliminary mapping experiments indicated that these gene complexes were
>150 kb apart, yet when MAT-1 (a1b1) and MAT-2 (a2b2)
strains were crossed, recombinant progeny with genotypes a1b2 and a2b1
were not found. Mating tests between parental strains and their progeny were
performed to search for these recombinant progeny (Bakkeren
and Kronstad, 1994).
We showed by tagging the a
and b gene complexes within both MAT loci with the recognition
sequence for the restriction enzyme I-SceI, that the MAT locus extends
over a large region and that the size and organization of the locus differs
between MAT-1 and MAT-2 strains. Specifically, we found that the
distance between the complexes is 500 kb in a MAT-1
strain and 430 kb in a MAT-2 strain. Characterization of the
organization of the known genes within the a and b gene complexes
provided evidence for non-homology and sequence inversion between MAT-1
and MAT-2. Antibiotic resistance markers were also used to tag the a
gene complex in MAT-1 strains (phleomycin) and the b gene complex
in MAT-2 strains (hygromycin). Crosses were performed with these
strains and progeny resistant to both antibiotics were recovered at a low
frequency suggesting that recombination is
suppressed within the MAT region. Overall, the chromosome homologues
carrying the U. hordei MAT locus share features with primitive sex
chromosomes (Lee et
al., 1999).
Recently, a BAC fingerprint map was constructed for the U. hordei genome and the complete 527 kb
MAT-1 region was sequenced.
Comparison with the respective U. maydis
chromosomal regions harbouring the a
and b mating-type complexes, revealed
synteny but also several translocations, indels, inversions and a large number
of transposon-like elements and repeats in U.
hordei (Bakkeren
et al., 2006). Comparison of mating-type regions and loci among
basidiomyctes reveales interesting evolutionary trends and points to the
generation of larger loci or even sex chromosomes (Bakkeren
et al., 2008).
Matings between related smut
species which do not occur naturally, can be forced by the introduction of
heterologous pheromone and cognate pheromone-receptor genes. This was
demonstrated between cells of two natural non-maters, U. hordei and U.
maydis (Bakkeren
and Kronstad, 1996).
Ustilago maydis
Many aspects of the mating
interactions, the molecular components involved and their downstream effectors,
targets and genes, as well as many aspects of pathogenicity and interaction
with host plants, have been studied in the model smut fungus, Ustilago
maydis, a close relative of U. hordei. For example, one of the key
features is the morphological switch from growth by budding of the
basidiospores to filamentous, pathogenic growth of the dikaryotic cell type
produced after mating. This process involves interesting kinase signal
transduction pathways. In addition, the complete genome of U. maydis has been sequenced and annotated (see Kamper et al, 2006;
accessible through the Broad
Institute website or the Munich
Information Centre for Protein Sequences). Several Ustilago
laboratories around the world are actively pursuing research on this
fungus.
Smutted
corn cobs or "huitlacoche",
are considered a delicacy in many Middle- and South-American countries.
Some other reviews for further
reading:
§ Feldbrugge,
M., J. Kamper, G. Steinberg and R. Kahmann (2004) Regulation of mating and
pathogenic development in Ustilago maydis.
Curr Opin Microbiol, 7: 666-672
- Kamper
et al (2006) Insights from the
genome of the biotrophic fungal plant pathogen Ustilago maydis. Nature,
444: 97-101
- Special
issue on U. maydis, August 2008.
Fungal Genetics & Biology, 45, Supplement 1, pp S1-S94
- Brefort, T., G. Doehlemann, et al. (2009). Ustilago maydis as a Pathogen. Ann. Rev. Phytopathol. 47: 423-445
Pathogenicity, virulence & avirulence
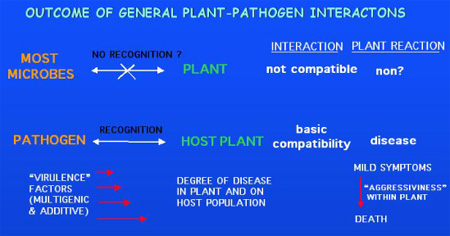
Many plant pathogens have been
shown genetically to harbour single dominant genes whose presence is recognized
by specific host cultivars possessing single, 'matching', dominant resistance
genes (‘gene-for-gene
interaction'). These pathogen genes have come to be known as avirulence or Avr
genes since recognition of their product by a host harbouring the cognate
resistance or R-gene (product) triggers a defence reaction (incompatible
interaction) which is epistatic over the potential of the pathogenic species to
infect the host (compatible interaction).

Much
research has focused on the understanding of the molecular workings of Avr
genes as well as their isolation because they a) represent single, often
dominant genes that are easy to track genetically, b) restrict host
range and c) are recognizable pathogenic factors that trigger host
defence. Defence mechanisms are often correlated with a conspicuous, necrotic
resistance reaction known as the hypersensitive response (HR), which is thought
to keep localized infections at bay. Initial responses during resistance but
also during compatible interactions, include a variety of molecular events in
both host and pathogen which are often the result of induced gene expression
(e.g., membrane depolarization, calcium influx, the production of reactive
oxygen species (ROS), pathogenesis related (PR-) proteins, etc.
Many
bacterial Avr genes have been isolated via classical bacterial genetic
techniques such as the transformation of a virulent receptor strain with a
genomic library from an avirulent strain. Avr-containing clones are
selected as those resulting in a visual hypersensitive response. Larger genomes
and inefficient transformation techniques have resulted in fewer such fungal
genes being isolated by similar methods. As a consequence, researchers have had
to resort to more complex techniques like reverse genetics and marker-assisted
genome walking. To date, a significant number of Avr genes have also been isolated from fungal and oomycete species,
aided by the analysis of total genome sequences of several model species. Many Avr gene products are small proteins
with signal peptides meant for secretion into plant host cells or apoplast
where most likely they fulfill a more-or-less essential (virulence or fitness)
function. In incompatible interactions they additionally and inadvertently
elicit defence responses. Mutation or deletion of the Avr genes yields avr alleles
which can escape recognition by the R-gene mediated surveillance system, but
might thereby invoke virulence or fitness penalties on its bearer. These Avr genes therefore presumably allow a
competitive advantage to the pathogen which seems logical in light of
evolutionary pressures. It follows that microbial “Avr” gene products likely have
been co-opted by some hosts as triggers for active defence. The molecular basis
for this type of resistance might be direct physical interaction of the Avr
gene product and the product of its cognate resistance gene (receptor-ligand
model), or indirectly with a host component in a complex over which an R-gene product stands guard (guard
hypothesis; reviewed in Bakkeren
and Gold, 2004, more recent: Jones and Dangl. 2006. The plant immune
system. Nature 444, 323-329).
U. hordei
avirulence genes
U. hordei displays
race-cultivar specialization. Fourteen races have been described which harbour
at least 5 single dominant Avr genes. In barley, a visible HR reaction
is not observed in an incompatible interaction. Therefore, currently,
incompatible interactions are scored after two to three months at heading by
the absence of disease symptoms in a significant number of plants compared to a
compatible control. A marker-based cloning approach for the isolation of Avr
genes was selected because a) although optimized, transformation is
still inefficient, b) the genome complexity is estimated at approx. 20
Mbp per haploid genome, making integration of random clones and subsequent
pathogenicity tests of at least three independent transformants on at least 50
plants each unfeasible and c) no race-specific elicitor has been found
to date making a reverse genetics approach impossible. As a first step a
virulent parent (MAT-2) was crossed with an avirulent parent (MAT-1)
on the universal susceptible barley cultivar ‘Odessa’, to construct a
population in which three unlinked Avr genes were segregating. Each
progeny was tested by back crosses on three differentials carrying cognate
resistance genes to test for Avr genotype. Marker analysis on genomic
DNAs from eight segregants pooled for Avr1 revealed one AFLP and two
RAPD markers. No positive clones were obtained from cosmid libraries
necessitating the construction of a Bacterial Artificial Chromosome (BAC)
library (Linning
et al., 2004). Biological activity tests of subclones require their
transformation into the virulent strain (MAT 2, v1), subsequent mating
of the transformants with a compatible, virulent strain (MAT-1, v1), and
inoculation on seedlings of barley cultivar ‘Hannchen’ (Ruh1). Transformants
that have received an active Avr1 gene will not produce any sori at
heading.
Current research projects:
Ø Cloning and characterization of Ustilago hordei avirulence
genes, mode of action
·
Using AFLP, SSR and RAPD marker technology on bulked progeny, Avr1, one of five known avirulence genes
of the barley smut fungus, Ustilago hordei, was located on BAC clones
and delineated on a 40-kb region (Linning
et al., 2004). Confirmation of the isolation of Avr1 by functional
analysis and characterization of the gene is in progress. Using the published U. maydis and U. hordei genome sequences, we are selecting other markers linked
to UhAvr6 which should also allow the isolation of this
avirulence gene. Future work will include the functional analysis of these
avirulence genes, their potential interaction with host factors and occurrence
of their alleles in different smut races, species and populations.
·
In collaboration with Drs. G. Scoles,
B. Rossnagel
and T. Grewal of the Dept. of Plant Sciences, University of Saskatchewan, barley
resistance gene, Ruh1, recognizing U. hordei Avr1, was identified and was
mapped to the short arm of chromosome 1 (7H) between markers iPgd1A and BCD129
on the ‘Harrington’/TR306 map (Grewal
et al., 2008). In addition, we might have revealed novel U. hordei Avr genes and cognate resistance genes
by using a large collection of defined new barley cultivars and germplasm.
Ø U. hordei genome sequencing and comparative analyses
among smuts
·
The generation of a U.
hordei BAC library, fingerprint map (in collaboration with Dr. J. W.
Kronstad, Michael Smith Laboratories, University of British Columbia,
Vancouver, BC, Canada), and the generation of BAC-end sequences (Bakkeren
et al., 2008), has laid the basis for the generation of the complete U. hordei genome sequence (Laurie et al., 2012), a collaboration with J.
Schirawski (Rheinisch-Westfälische
Technische Hochschule Aachen University, Germany), R. Kahmann (Max Planck
Institute for Terrestrial
Microbiology, Marburg, Germany) and Drs. G. Mannhaupt, M.
Muensterkoetter, U. Gueldener and P. Wong at the
Münich Information center for Protein Sequences (MIPS,
Helmholtz Zentrum, München, Germany). Comparative analyses among the three,
curently generated smut genomes (U.
maydis, U. hordei and Sporisorium reilianum), with emphasis on
the diversity of effector genes, is in progress.
Ø
Barley host
responses during compatible and incompatible interactions
·
Microscopy and gene expression patterns,
isolation of involved genes. The isolation of an avirulence gene has
allowed us to construct isogenic fungal strains differing only in the absence
or presence of individual avirulence genes. Such strains permit the systematic
study of the role(s) these avirulence genes play in compatible versus
incompatible interactions. In incompatible interactions, no (visible)
hypersensitive response (HR) is generated but resistance is nevertheless
achieved through arrest of fungal hyphal growth. Preliminary data suggest that
different Avr / R-gene
combinations "act" at different stages during pathogen invasion. The Avr1-Ruh1 interaction was studied in
detail on the microscopic level; a localized, microscopic HR takes place
immediately upon penetration of the first cell layer of the barley seedling,
including collapse of few cells surrounding the penetration site (Hu et
al., 2003). This data has aided in selecting the timing and tissues for RNA
isolation for transcript profiling of the barley host response using
microarrays. Parallel
work on the defence and resistance pathways in wheat and barley is being
carried out. Similarly, nonhost interactions have been
studied by applying U. hordei to
wheat & Triticale (Gaudet
et al., 2010; collaboration with Drs. D. Gaudet and A. Laroche,
Lethbridge Research Centre, Lethbridge, AB).